Merge data at different TMT plexes
The standard Tandem Mass Tag@ (TMT) kits are capable of relative quantitation with up to 11 samples under one experiment, by adding a chemical structure at a monoisotopic mass of 229.162932 Da to the lysine residues or the N-terminals of peptides. More recently, TMTpro was made available for the simultaneous quantitation with up to 16 samples in one experiment.
TMTpro is similar in design to the standard TMT, but will tag the amine groups in peptides with a monoisotopic mass of 304.207146 Da. Due to the difference in precursor masses between TMT and TMTpro, we cannot simply perform a database search for standard TMT data using TMTpro settings.
Chances may remain that we were allured to put together old data from standard TMT experiments and new data from TMTpro experiments.1 We might simply perform database searches against TMT and TMTpro data with separate sets of parameters. However, the so-generated PSM outputs will be different in both the numbers and the keys of columns.
One resort is to manually compile PSM files to the same column format. Alternatively, we can let proteoQ automate the process by padding additional TMT channels in PSM outputs. In the hypothetical example shown below, we have two sets of TMT data, one at 6-plex and the other at 16-plex. We simply provide the sample information in a metadata file (named expt_smry.xlsx by default):
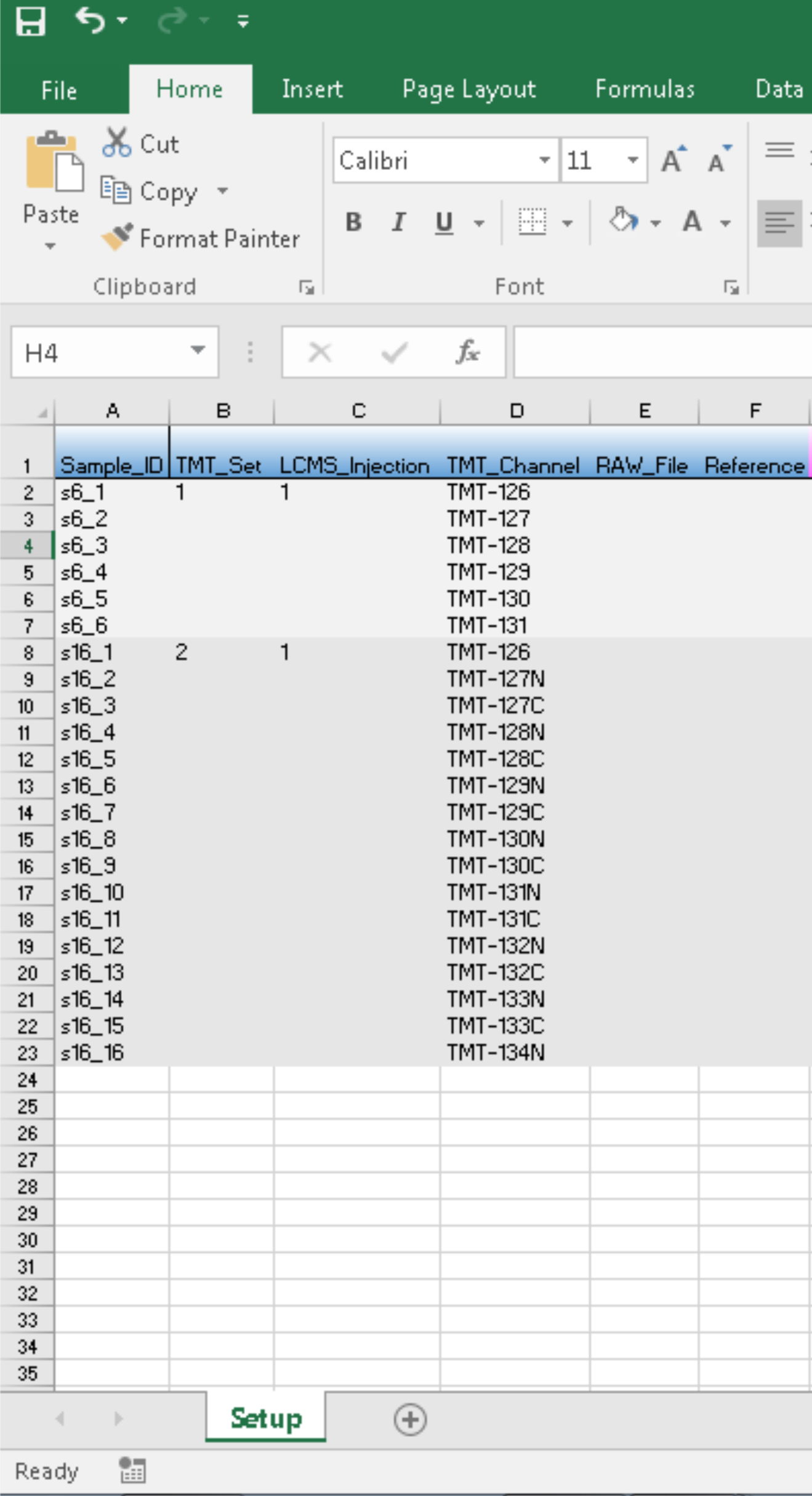
and load the experiment via load_expts(). The updated expt_smry.xlsx will look like:
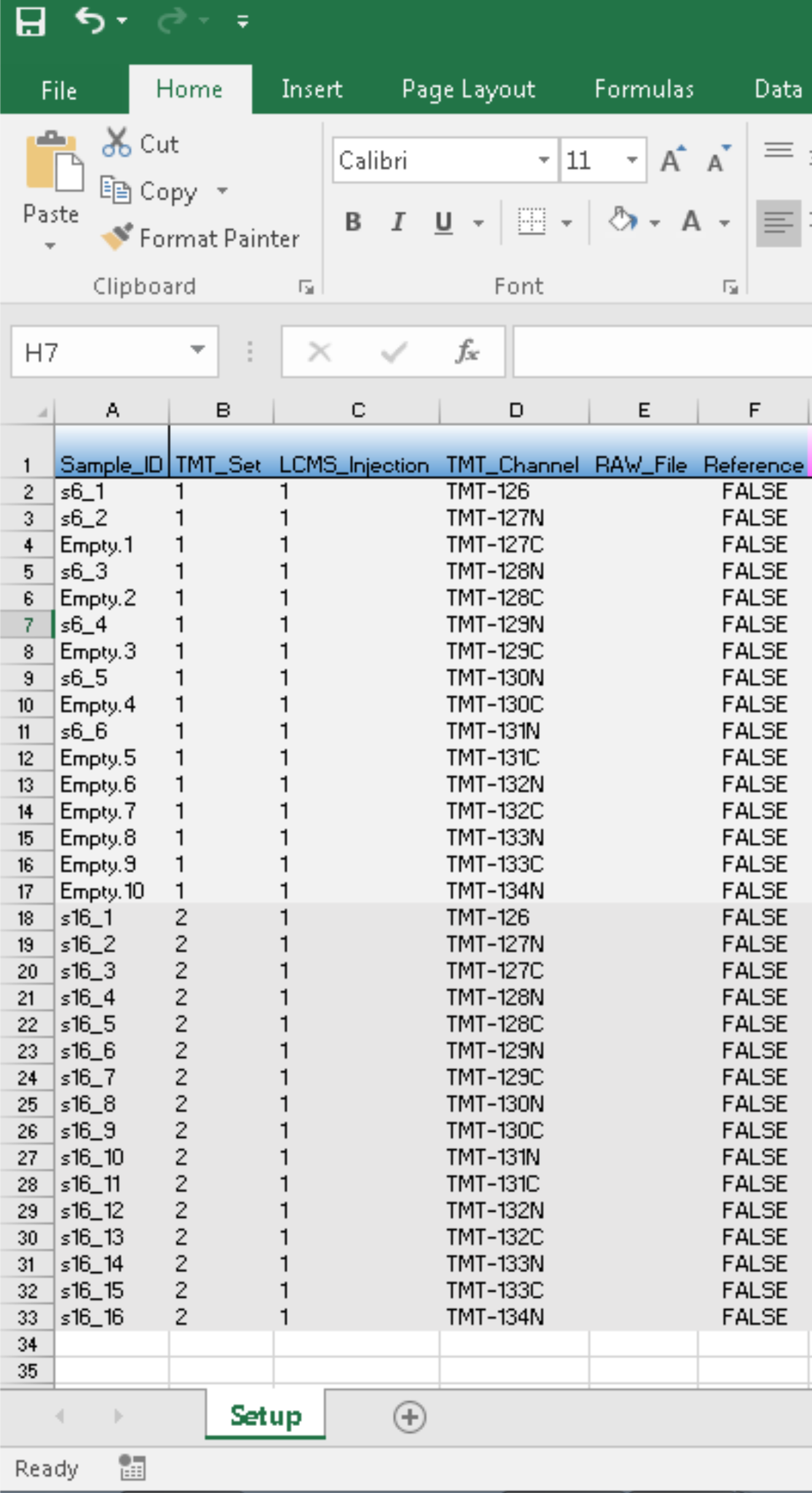
The proteoQ will later take the standardized metadata for padding the PSM table from 6-plex TMT to the highest possible plex, which is 16-plex, in this example.
In a next post, I will show how to utilize repetitively the same RAW MS file with proteoQ. For example, one piece of MS file may be searched with different sets of parameters and analyzed side-by-side.
Of course, we need to apply the same reference material and be comfortable with the assumption of reliable peptide references over time↩︎
Qiang Zhang
Instructor of Medicine
My research interests include mass spectrometry-based proteomics and automation in data analysis.