Sample exclusions
With proteoQ, the removal of sample(s) from analysis is a simple matter of deleting corresponding entries under column Sample_ID in a metadata file.
In the example shown below, we choose to exclude the two samples at TMT channels of 129N and 130C from the experiment at TMT_Set 1:
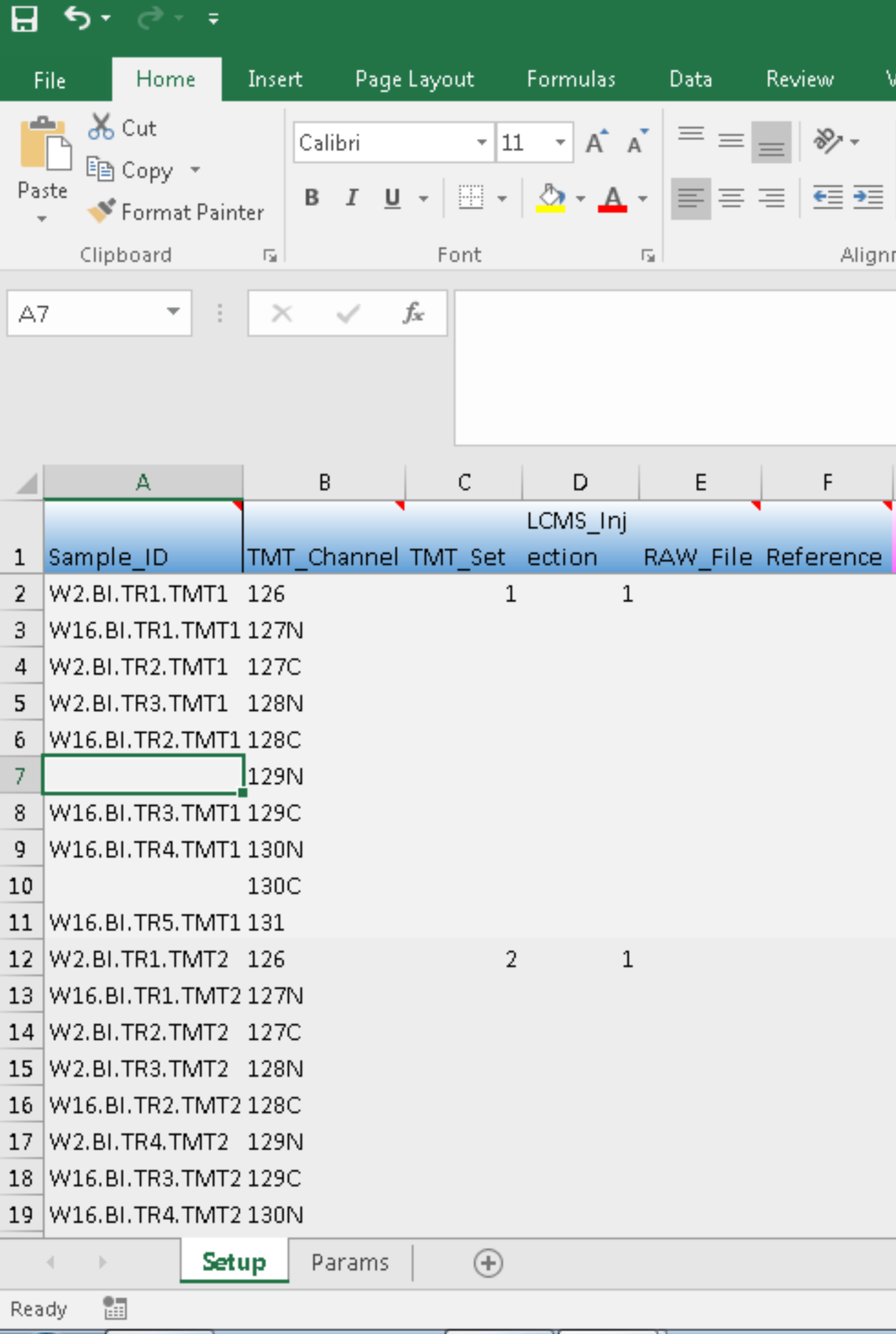
we then process the metadata after saving the changes that we have made:
library(proteoQ)
load_expts("~\\proteoQ\\examples", "expt_smry.xlsx")We can see that the two sample entries for tentative removals are now named Empty.1 and Empty.2.
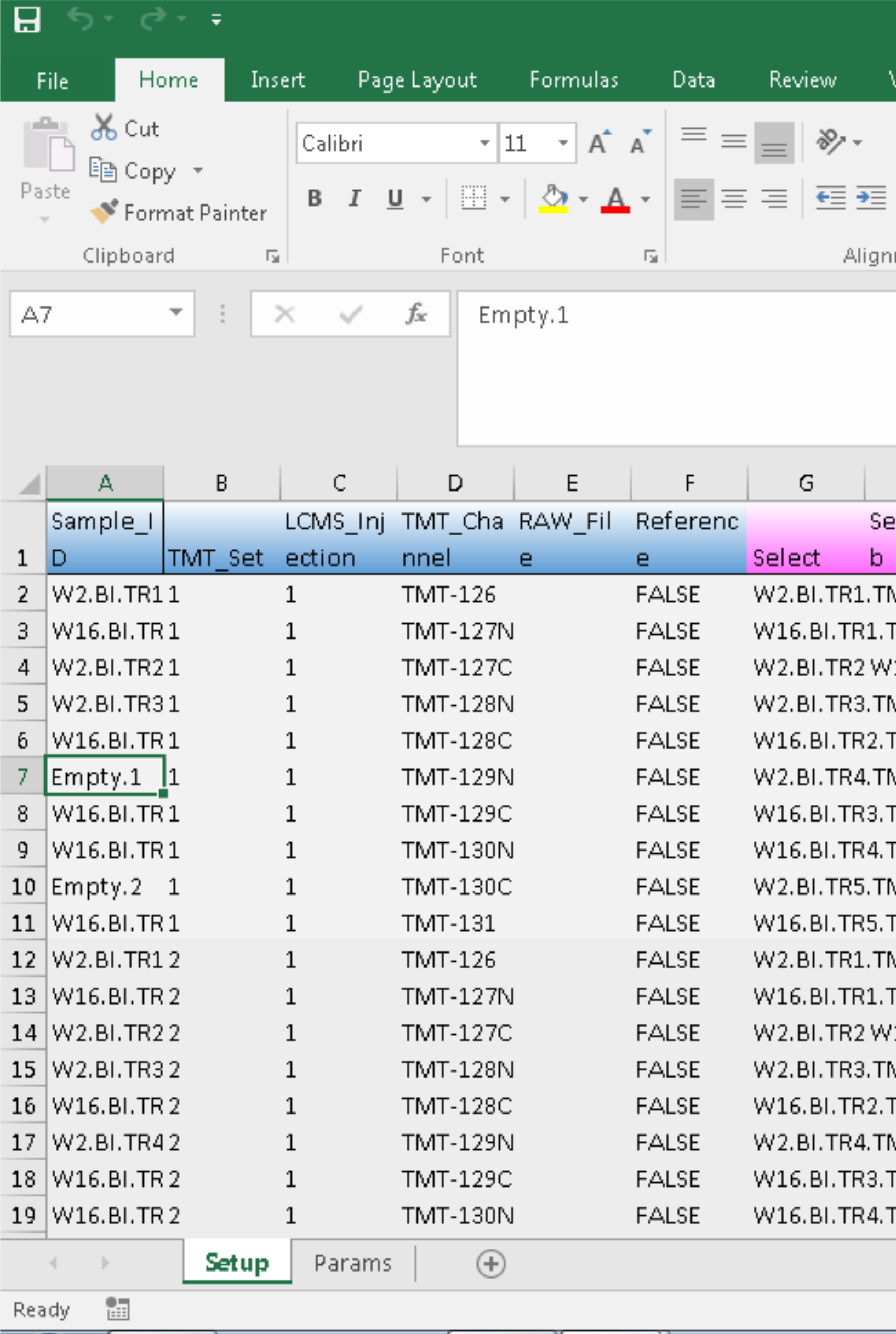
As you may tell, the character strings of Empty.[...] are reserved in proteoQ for samples that will not be processed.
Following the updates in metadata, the entire workflow starting from normPSM needs to be re-executed. The essence of re-execution is to update files of PSM, peptide and protein tables, as well as various informatic findings. This process will modify broadly the outputs that residue previously under a given working directory. It is therefore recommended that the re-analysis being treated as brand new and assessed under a freshly made file folder.
It is also possible to delete the entire row(s) in metadata for sample removals. This approach might have an additional special use where we would have to join data at different TMT multiplicity, for example, combining data from 10-plex and 11-plex experiments. I will discuss in details in a next post.
Qiang Zhang
Instructor of Medicine
My research interests include mass spectrometry-based proteomics and automation in data analysis.