Metadata files for LFQ
The LFQ workflows in proteoQ take the same metadata files as those with TMT procedures; namely, expt_smry.xlsx and frac_smry.xlsx by default.
No prefractionation
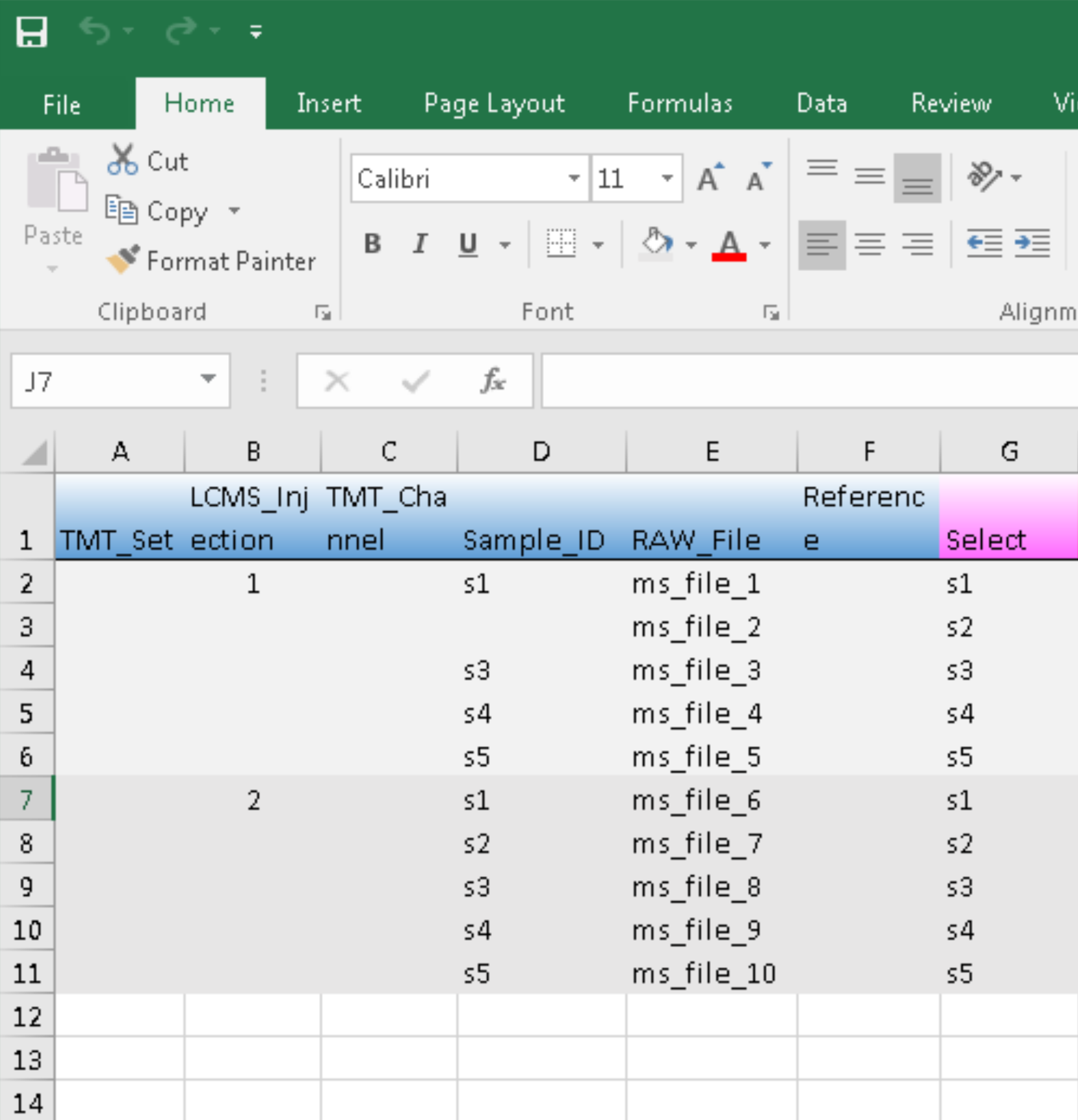
For experiments without peptide prefractionation, we only need to prepare the expt_smry.xlsx file, with values under column TMT_Set and TMT_Channel being left blank:

Analogous to TMT experiments, we may allocate certain samples to be “references” by entering no-empty characters in the corresponding cells under column Reference.
To exclude samples from analysis, we may delete the respective entries under column Sample_ID. In the example shown below, we deleted the entry of sample s2 from LCMS series 1 (see also an earlier post of sample exlcusion).
With prefractionation
For data with peptide prefractionation, we would need to empty the entries under column RAW_File in expt_smry.xlsx and entered the extended file names in frac_smry.xslx:
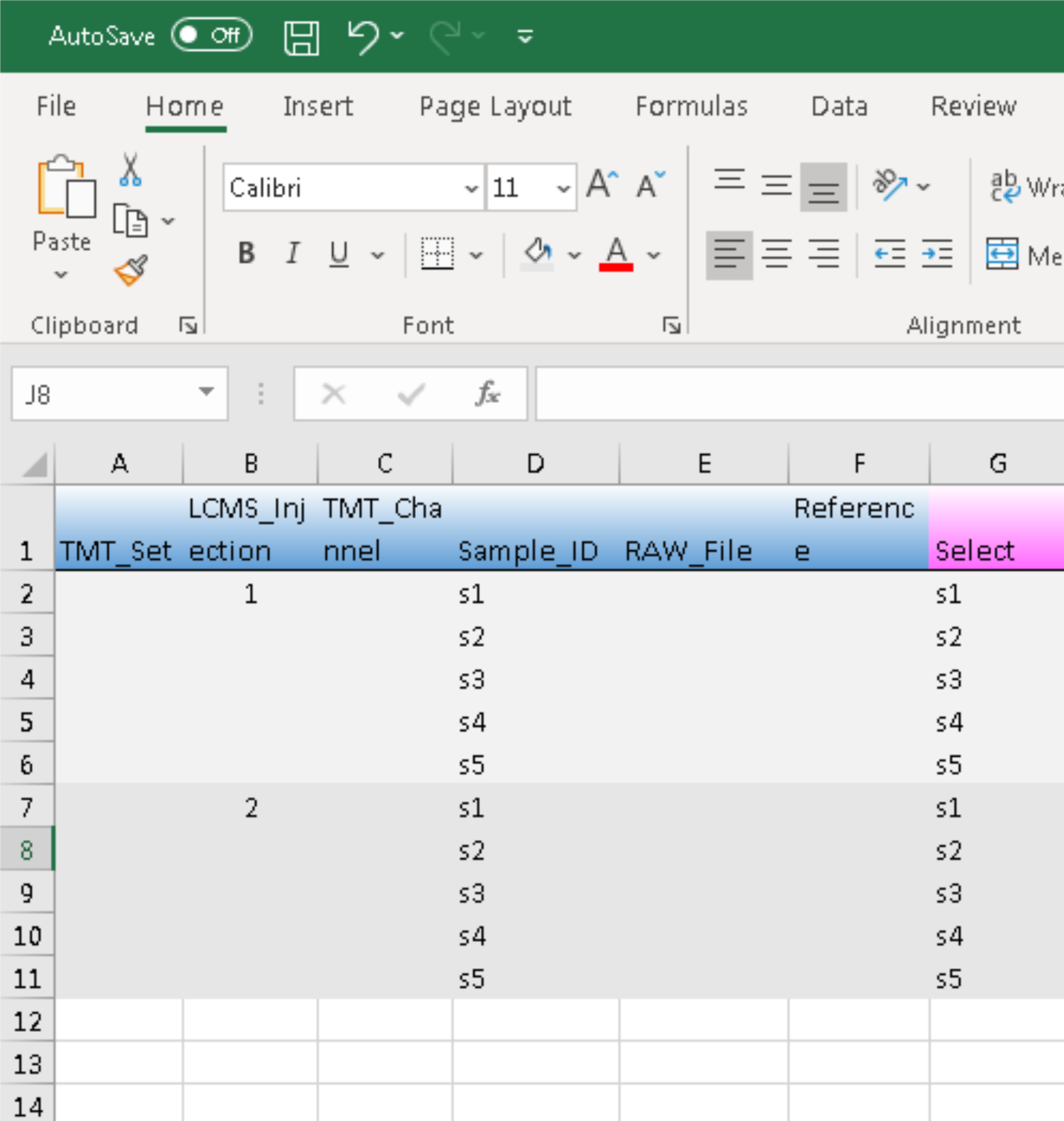
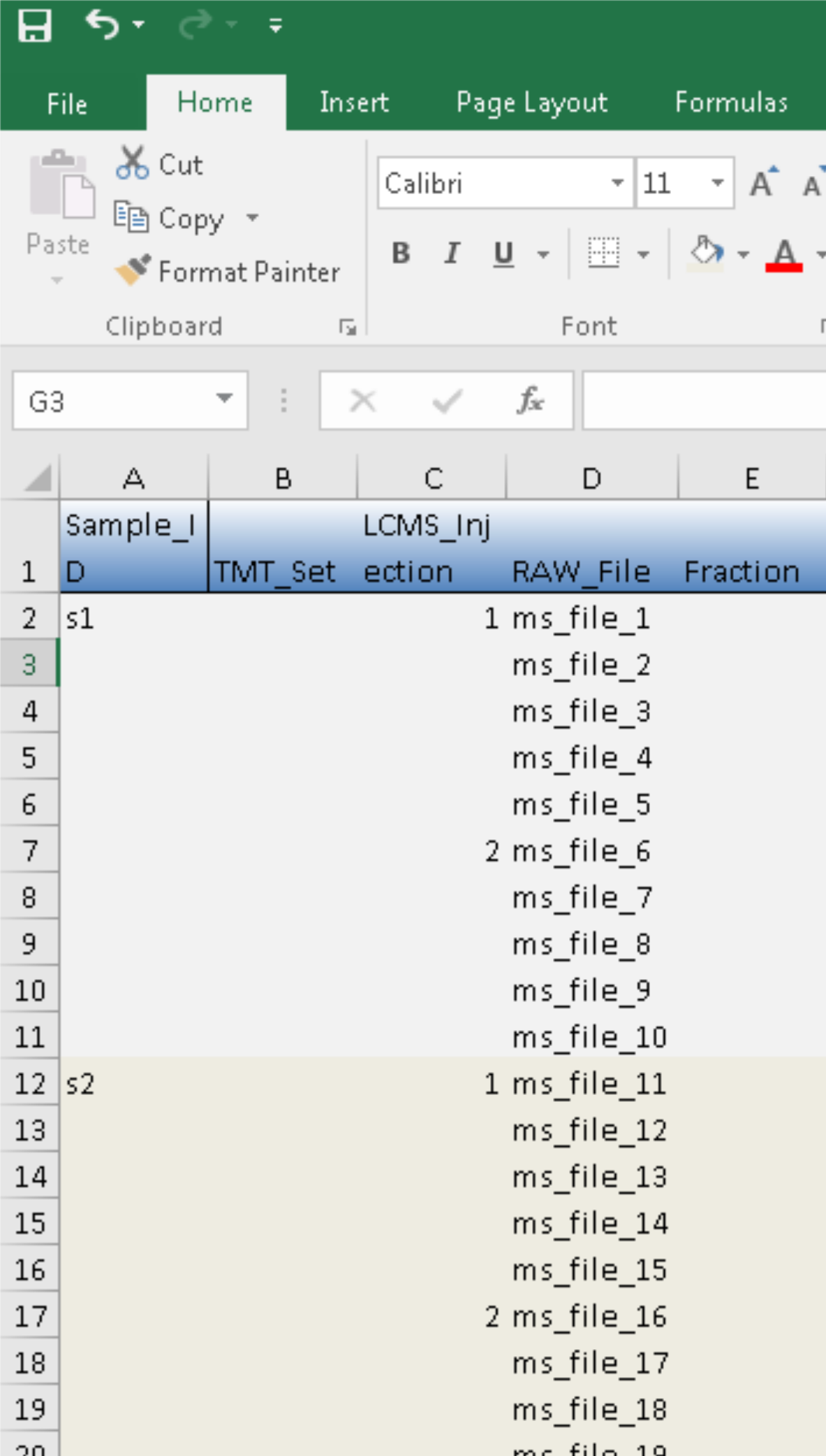
This is the same as preparing TMT metadata. However, there is an additional column of Sample_ID in the LFQ template. It will be used to match those in expt_smry.xlsx.
Difference to TMT
Note that the utility proteoQ::purgePSM will have no effect with LFQ workflows as peak areas/intensity from extracted-ion chromatograms are typically employed in LFQ. In other words, multiple PSMs at the same vicinity of LC retention time would be used to construct the same (single) peak profile at MS1 levels for quantitation. More details for the topic are available in the help documents of proteoQ via ?load_expts.
Qiang Zhang
Instructor of Medicine
My research interests include mass spectrometry-based proteomics and automation in data analysis.